Inputs / Outputs¶
craw_coverage¶
Inputs¶
craw_coverage* need a file bam or wig to compute coverage and an annotation file to specify on which regions to compute these coverages.
bam file¶
craw_coverage can use a file of alignment reads called bam file. a bam file is a short DNA sequence read alignments in the Binary Alignment/Map format (.bam). craw_coverage needs also the corresponding index file (bai). The index file must be located beside the bam file with the same name instead to have the .bam extension it end by .bai extension. If you have not the index file you have to create it.
To index a bam file you need samtools. The command line is
samtools index file.bam
For more explanation see http://www.htslib.org/doc/ .
wig file¶
craw_coverage can compute coverage also from wig file see https://wiki.nci.nih.gov/display/tcga/wiggle+format+specification and http://genome.ucsc.edu/goldenPath/help/wiggle.html . for format specifications. Compare d to these specifications craw support coverages on both strands. the positive coverages scores are on the forward strand whereas the negative ones are on the reverse strand.
track type=wiggle_0 name="demo" color=96,144,246 altColor=96,144,246 autoScale=on graphType=bar
variableStep chrom=chrI span=1
72 12.0000
73 35.0000
74 70.0000
75 127.0000
...
72 -88.0000
73 -42.0000
74 -12.0000
75 -1.0000
In the example above the coverage on the Chromosome I for the positions 72, 73, 74, 75 are 12, 35, 70, 127 on the forward strand and 88, 42, 12, 1 on the reverse strand.
annotation file¶
The annotation file is a tsv file by default. It’s mean that it is a text file with value separated by tabulation (not spaces) or commas. But if a separator is specified (–sep) it can be a csv file or any columns file.
The first line of the file must be the name of the columns the other lines the values. Each line represent a row.
name gene chromosome strand Position
YEL072W RMD6 chrV + 14415
YEL071W DLD3 chrV + 17845
YEL070W DSF1 chrV + 21097
YEL066W HPA3 chrV + 27206
YEL065W SIT1 chrV + 29543
YEL062W NPR2 chrV + 36254
YEL058W PCM1 chrV + 44925
YEL056W HAT2 chrV + 48373
All lines starting with ‘#’ character will be ignored.
# This is the annotation file for Wild type
# bla bla ...
name gene chromosome strand Position
YEL072W RMD6 chrV + 14415
YEL071W DLD3 chrV + 17845
YEL070W DSF1 chrV + 21097
YEL066W HPA3 chrV + 27206
YEL065W SIT1 chrV + 29543
YEL062W NPR2 chrV + 36254
YEL058W PCM1 chrV + 44925
YEL056W HAT2 chrV + 48373
mandatory columns¶
There is 3 mandatory columns in the annotation file.
columns with fixed name¶
two with a fixed name:
strand indicate on which strand is located the region of interest. The authorized values for this columns are +/- , 1/-1 or for/rev.
chromosome the chromosome name where is located the region of interest.
columns with variable name¶
In addition of these two columns the column to define the position of reference is mandatory too, but the name of this column can be specified by the user. If it’s not craw_coverage will use a column name ‘position’.
If we want to compute coverage on variable window size, 2 extra columns whose name must be specified by the user by the following option:
–start-col to define the beginning of the window (this position is included in the window)
–stop-col to define the end of the window (this position is included in the window)
name gene type chromosome strand annotation_start annotation_end has_transcript transcription_end transcription_start
YEL072W RMD6 gene chrV 1 13720 14415 1 14745 13569
YEL071W DLD3 gene chrV 1 16355 17845 1 17881 16177
YEL070W DSF1 gene chrV 1 19589 21097 1 21197 19539
YEL066W HPA3 gene chrV 1 26721 27206 1 27625 26137
YEL065W SIT1 gene chrV 1 27657 29543 1 29601 27625
YEL062W NPR2 gene chrV 1 34407 36254 1 36401 34321
YEL058W PCM1 gene chrV 1 43252 44925 1 44993 43217
YEL056W HAT2 gene chrV 1 47168 48373 1 48457 47105
YEL052W AFG1 gene chrV 1 56571 58100 1 58105 56537
craw_coverage --wig file.wig --annot annot.txt --ref-col annotation_start --start-col annotation_start --stop-col annotation_end
The position of reference must be between start and end. The authorized values are positive integers.
Note
the position of reference can be used to define the reference and the start ot the end of the window.
craw_coverage --bam file.bam --annot annot.txt --ref-col annotation_start --start-col annotation_start --stop-col annotation_end
All other columns are not necessary but will be reported as is in the coverage file.
Outputs¶
coverage_file¶
It’s a tsv file with all columns found in annotation file plus the result of coverage position by position centered on the reference position define for each line. for instance
craw_coverage --wig=../data/small.wig --annot=../data/annotations.txt
--ref-col=annotation_start --before=0 --after=2000
In the command line above, the column ‘0’ correspond to the annotation_start position the column ‘1’ to annotation_start + 1 on so on until ‘2000’ (here we display only the first 3 columns of the coverage).
# Running Counter RnAseq Window craw_coverage
# Version: craw NOT packaged, it should be a development version | Python 3.4
# Using: pysam 0.9.1.4 (samtools 1.3.1)
#
# craw_coverage run with the following arguments:
# --after=3
# --annot=../data/annotation_wo_start.txt
# --before=5
# --chr-col=chromosome
# --output=small_wig.cov
# --qual-thr=0
# --quiet=1
# --ref-col=Position
# --sense=mixed
# --sep=
# --strand-col=strand
# --suffix=cov
# --verbose=0
# --wig=../data/small.wig
sense name gene type chromosome strand annotation_start annotation_end has_transcript transcription_end transcription_start 0 1 2
S YEL072W RMD6 gene chrV + 13720 14415 1 14745 13569 7 7 7
AS YEL072W RMD6 gene chrV + 13720 14415 1 14745 13569 0 0 0
S YEL071W DLD3 gene chrV + 16355 17845 1 17881 16177 31 33 33
The line starting with ‘#’ are comments and will be ignored for further processing. But in traceability/reproducibility concern, in the comments craw_coverage indicate the version of the program and the arguments used for this experiment.
craw_htmp¶
Outputs¶
The default output of craw_htmp (if –out is omitted) is grapical window on the screen. The figure display on the screen can be saved using the window menu.
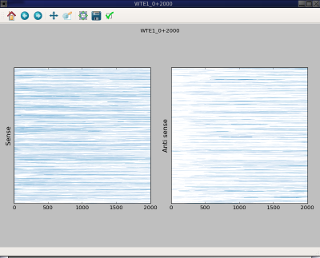
It is also possible to generate directly a image file in various format by specifying the –out option. The output format will be deduced form the filename extension provide to –out option.
--out foo.jpeg for jpeg image or --out foo.png for png image
The supported format vary in function of the matloblib backend used (see matplotlib configuration).
If –size raw is used 2 files will be generated one for the sense and the other for the antisense. If –out is not specified it will be the name of the coverage file without extension and the format will be png.
craw_htmp foo_bar.cov --size raw
will produce foo_bar.sense.png and foo_bar.antisense.png
craw_htmp foo_bar.cov --size raw --out Xyzzy.jpeg
will produce Xyzzy.sense.jpeg and Xyzzy.antisense.jpeg