overview¶
Counter RNA seq Window is a package which aim to compute and visualize the coverage of RNA seq experiment.
The craw package contains two scripts craw_coverage and craw_htmp. craw_coverage compute the coverage, whereas craw_htmp allow to represent graphically the results of craw_coverage with a heat map.
craw_coverage¶
craw_coverage take as input a bam file or wig file and an annotation file. The annotation file describe on which gene the craw_coverage must compute the coverage. The script compute a coverage for each position of this gene on a specified window around a position of reference on both sense and put the results on a matrix. The region of interest can be fixed for all genes (specified by the command line) or variable. In the this case the annotation file must contains two columns to specify beginning and the end of the region to take in account. The results in the matrix are centered on the position of reference of each gene. In the case of variable length of window the results are padded on left and right if necessary with None value. The results is saved in a file as a tabulated separated value by default with the same name as the bam file with the .cov extension (see Outputs for more details).
Below an example to illustrate how craw_coverage work. If we consider the following genome and we want to analyze 3 gene foo, bar, buz
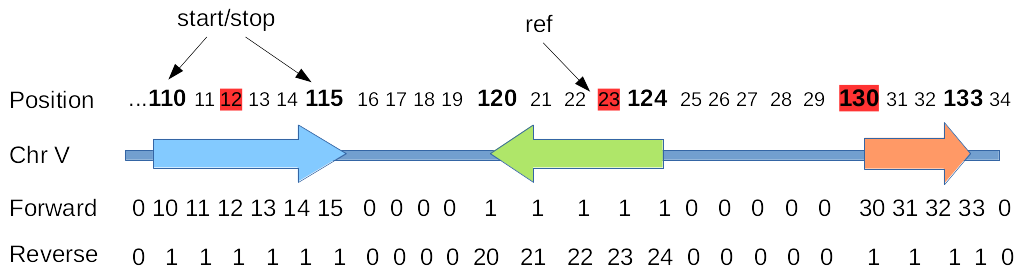
On the figure above
The first line represent the positions on the genome (1-based)
The bold position indicate the boundaries of region we want to analyse.
the red highlighted positions indicate, for each region, the position of reference.
the second line represent the genes and their respective sense.
the 2 last lines the coverage at each position of the genome for each strand
So to analyse these genes, we create an annotation file like following.
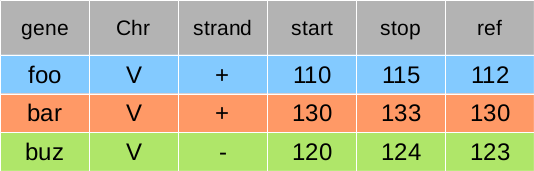
the run a command line like:
craw_coverage --bam mygenome.bam --annot my_annotation --ref-col ref --start-col start --stop-col stop
will produce the following coverage matrix
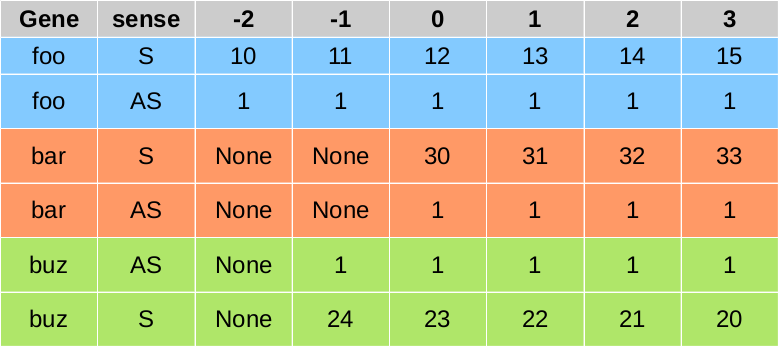
craw_htmp¶
craw_htmp read coverage file produced by craw_coverage and generate a graphical representation. It can produce either a file or an interactive graphic. The look and feel of the graphic and the format of supported outputs vary in function of the backend of matplotlib used (see matplotlib configuration ). It can also produce raw images using pillow where 1 nucleotide is represent by 1 pixel.
Licensing¶
All files belonging to the Counter RNAseqWindow (craw) package. are distributed under the GPLv3 licensing.
You should have received a copy of the GNU General Public License along with the package (see COPYING file). If not, see <http://www.gnu.org/licenses/>.
craw is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version.
craw is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
Authors: Bertrand Neron Copyright © 2017-2019 Institut Pasteur (Paris). see COPYRIGHT file for details.